
Endotoxin Control Strategies for Parenteral Drug Product Manufacturing
Posted on July 13, 2018
Introduction
Control of bacterial endotoxins, gram-negative bacteria that can cause pyrogenicity, is critical in the manufacture of pharmaceutical drug products intended for parenteral administration. Unlike bioburden, which can be readily eliminated by terminal sterilization via autoclave or filtration through a 0.22 µm membrane during aseptic filling, bacterial endotoxin is difficult to remove during routine manufacturing operations. As such, it is essential to implement proper endotoxin controls at every point of potential introduction in the manufacturing process.
Endotoxin control is especially relevant for therapeutics intended for either high dosing volumes or patients with low body weights. United States Pharmacopeia <85>1 defines the limits for parenteral drugs as:
Endotoxin limit = K/M
where K is the threshold dose of endotoxin per kg of body weight and M is the maximum bolus dose per kg of body weight. For most parenteral routes of administration, such as those administered intravenously or subcutaneously, K is defined as 5 EU/kg of body weight. This is then adjusted based on the bolus dose size and patient weight to calculate a final endotoxin limit. As the bolus dose size is decreased or the patient size is increased, the endotoxin limit is increased.
Intrathecal injection is a route of administration that offers the unique advantage of providing direct access to the central nervous system and the brain without the need to pass the blood brain barrier. When calculating the endotoxin limit for therapeutics given via the intrathecal route of administration, K is significantly reduced to 0.2 EU/kg of body weight, resulting in a significantly lower endotoxin limit in the final drug product. Should the intended patient population be neonatal or pediatric, these limits can be decreased even further as a function of patient weight. Finally, if a larger dose volume is required, the limit is reduced further. In some cases, this limit can approach or possibly even surpass that of Water for Injection (WFI), typically <0.25 EU/mL.
The following summary provides an end-to-end approach to develop an appropriate endotoxin control strategy for a typical parenteral drug product manufacturing process. Initially, all potential sources of endotoxin are identified. The risk of each of these sources is categorized relative to the total allowable endotoxin in the final drug product. A stacked tolerance analysis is performed to assess the collective risk from all the identified sources to the final endotoxin limit. Risk-based mitigation strategies relying upon historical testing results, incoming goods controls and vendor specifications are implemented to ensure a final drug product that can be reliably manufactured to meet the required endotoxin limit.
Potential Sources of Endotoxin
The first step in developing an endotoxin control strategy is to identify all the potential points in the manufacturing process where endotoxin may be introduced. For the purposes of this discussion, a routine manufacturing process will be considered where an active pharmaceutical ingredient (API) powder is converted into a final liquid drug product intended for parenteral administration (Figure 1).

In this process, a buffer is prepared in the compounding vessel by dissolving a series of excipients in WFI. The API is reconstituted in a separate container and compounded with the prepared buffer in the vessel before WFI is added to dilute to the final target volume to create the bulk drug product. The resulting bulk drug product solution is then passed through a bioburden reduction filter into a holding vessel to reduce the chance of microbial growth during any manufacturing hold time prior to the final filling steps. Aseptic filling is then conducted in a grade A space by passing the bulk drug product through sterilizing grade 0.2 µm filters. Figure 1 depicts the filling being performed into vials which are stoppered and capped for a final liquid drug product presentation. However, the drug product could also undergo a freeze-drying process step to result in a lyophilized drug product. Alternatively, the primary containers for the final drug product could be syringes or cartridges instead of vials.
Potential sources of endotoxin include any material that is present in the final drug product presentation or that may come in contact with the drug product during manufacturing. These sources can be grouped into three categories; raw materials, process equipment and container closure components. In assessing the overall endotoxin risk of the manufacturing process, not only must each potential endotoxin source be identified, but the theoretical maximum endotoxin from each source needs to be calculated. This can only be done by understanding the endotoxin controls or specifications for each individual source.
One significant potential source of endotoxin is the raw materials. In this case, raw materials may include anything present in the final bulk drug product, specifically the API, WFI and any excipients. Manufacturing sites have an internal specification and routine testing regiments for endotoxin on the WFI lines. This internal specification can be used as the theoretical worst-case contribution of the WFI to the final drug product.
The maximum potential endotoxin introduction from the API can be calculated using the endotoxin limit for the API and the amount of API that is included in a single drug product dose. A similar calculation can be conducted for each excipient using the endotoxin specification found on the certificate of analysis from the supplier. In some cases, vendors do not conduct endotoxin testing on a particular excipient. Thus, for drug products that must meet a low final endotoxin limit, it is critical to ensure that excipients are sourced from vendors that test and report endotoxin for their products. Alternatively further incoming goods testing may be conducted by the manufacturing site.
A typical manufacturing process includes an array of components such as silicone tubing, filing needles and filtration equipment. Additionally, vessels used for the compounding of the bulk drug product can either be disposable bags composed of plastics or reusable stainless steel vessels. An assessment of the endotoxin risk posed by a piece of disposable equipment can be challenging. This equipment is provided by the vendor with an endotoxin specification that can be found on the certificate of analysis. While this value is readily available, the interpretation of the overall risk to the final drug product requires knowledge about the testing method used by the vendor. To measure endotoxin, a typical testing program will soak a smaller piece of the equipment in endotoxin free water for a specified amount of time. This is followed by measuring the endotoxin in the water in order to determine the amount that has been introduced from the piece of equipment. Thus, in order to assess the overall risk to the drug product manufacturing process, one needs to know the volume of endotoxin free water used in the soak, the size of the component tested and the fraction of the surface area of the component in contact with the water, in order to appropriately scale the potential endotoxin to the equipment used in the manufacturing process.
For example, as an illustration of the calculation, consider silicone tubing that has an endotoxin specification of <0.5 EU/mL. If the test method involves soaking a 1 meter piece of tubing in 500 mL of endotoxin free water, it can be calculated that the highest amount of endotoxin present on the test piece of tubing for it to still pass specification would be 250 EU. This can then be scaled to the amount of tubing used in the manufacturing process, divided by the batch size and multiplied by the final drug product dose in order to calculate the maximum theoretical contribution of endotoxin from that individual component. The challenge in conducting this specific component analysis is that the endotoxin test methods are often confidential and not easily obtainable from vendors without a quality audit. Thus, in order to conduct such an assessment, it is critical for manufacturers and vendors to work collaboratively in order to ensure the componentry selected for use is appropriate for the process that is being implemented.
For non-disposable equipment used in the process, such as stainless steel compounding vessels and filling needles, potential endotoxin risks are generally evaluated during the cleaning validation studies at the manufacturing site. Bioburden and endotoxin samples should be collected and tested against specifications at the end of the washing cycle after a “dirty hold time” challenge, outside of the normally allowed time limit prior to cleaning following manufacturing. In cases where endotoxin limits in the drug product are very stringent, the use of a caustic cleaning agent is recommended in the cleaning cycle. Caustic solutions are known to degrade and eliminate endotoxin even if the product does not require their use for the elimination of product carryover. Worst case cleaning locations are selected based on a risk analysis of difficult to clean areas, for example inside of J-tubes, tank outlet drains, impeller fins, etc. While these locations are assessed for product removal and carryover calculations, they are typically not swabbed for endotoxin. These locations could be swabbed and tested in the case of products with low tolerable levels of endotoxin in the final product to add additional endotoxin removal assurance if required.
Finally, primary packaging components are another potential source of endotoxin. For a liquid or lyophilized drug product in a vial, endotoxin may be introduced by either the vial or the stopper. Typically endotoxin introduced into the product from the vial is not of significant concern. Aseptically filled vials are routinely subjected to depyrogenation/dry-heat sterilization prior to filling as part of the manufacturing process using a dry heat tunnel downstream from the vial washer that forms the barrier with the Grade A/Class 100 sterile processing area. The expectation outlined in USP <1211>2 and European Pharmacopeia Chapter 9.13 is that dry heat sterilization process be validated to remove not less than 3 logs of endotoxin. The kinetics of pyrogen deactivation have been studied extensively4 with higher temperatures requiring dramatically shorter time for 3 log endotoxin removal. The temperature setpoint and the residence time at temperature (or heat tunnel belt speed) should be controlled during aseptic processing. During initial validation of the equipment and during yearly requalification the worst-case (lowest) temperature setpoint and minimum allowable residence time should be challenged while measuring vial temperatures across the hot zone and sampling for endotoxin levels prior to and after the depyrogenation cycle to confirm 3 log endotoxin removal.
Modern, ready to sterilize stoppers do not usually undergo any type of preparation at the manufacturing site that will result in the reduction of endotoxin. Rubber components such as vial stoppers cannot withstand typical depyrogenation temperatures and are generally sterilized using moist heat in an autoclave which does not remove endotoxin. Thus, incoming goods testing and vendor specifications are the critical control point for ensuring endotoxin control of these components. Specific attention needs to be paid to the endotoxin removal during the validation of the washing process at the stopper manufacturer or stopper processing site. To assess the potential risk to the product, the endotoxin specification can be divided by the dose volume to determine the maximum theoretical contribution to the final drug product. A similar assessment can be done for ready to fill syringes or cartridges if they are used as the drug product presentation. Special attention should be given if those devices are not received at the manufacturing site in a ready to fill configuration. The required processing and assembly operations at the manufacturing site should be scrutinized to ensure this is performed in a manner which does not introduce endotoxin into the product.
The endotoxin sources considered here are all physical components of either the final drug product presentation or the manufacturing process. It should be noted that good aseptic manufacturing practices by site personnel are critical to ensure a final drug product with low endotoxin. In general, if these practices are well established and followed, the risk of endotoxin introduction by manufacturing personnel can be considered low. Thorough assessments of a site’s aseptic handling practices are regularly conducted during routine quality audits and inspections. These audits should ensure that the acceptance criteria used for facility assessments such as media fill reviews and washing and preparation of tools are suitable to account for the needed endotoxin requirements of the final drug product.
Stack Tolerance Risk Assessment and Mitigation Strategies
Once all possible sources of endotoxin have been identified and the theoretical maximum contribution for each has been determined, a control strategy can be developed. Figure 2 provides a flowchart to assess the need for additional control points to ensure the final drug product will meet the specification. Each potential source of endotoxin is categorized by its maximum potential contribution. Sources which may introduce enough endotoxin that the final drug product would fail release testing if this individual source met its theoretical maximum are categorized as high risk components. Low risk components are those that in a worst case scenario would not introduce enough endotoxin to exceed 5% of the drug product limit. Sources that could provide an intermediate level between 5% and 100% of the final drug product endotoxin limit are deemed moderate risk.
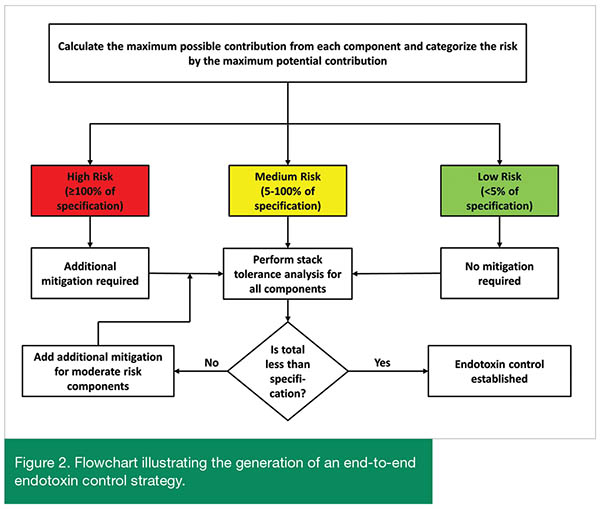
Mitigation of endotoxin introduction begins with the high risk components. Control of endotoxin in a manufacturing process cannot be claimed if a single component in that process can introduce enough endotoxin to result in the failure of the drug product to meet the endotoxin specification. Thus, before an end-to-end assessment of endotoxin risk is conducted, further process controls need to be implemented for those components which have been identified as high risk. After these additional mitigations have been established, then a stack tolerance can be performed to assess the overall end-to-end risk from the entire manufacturing process. In the stack tolerance analysis, the maximum potential endotoxin contribution from all potential sources, regardless of risk category, are added together. This total represents the worst-case value for endotoxin in the final drug product. If this value is lower than the drug product endotoxin specification, control during the manufacturing process can be claimed. If the value is higher than the specification, additional process controls and mitigations need to be implemented in order to ensure the final drug product will reliably pass the specification.
There are several mitigation strategies available to decrease the overall risk of endotoxin introduction during the manufacturing process. The most direct point of additional control is during incoming goods inspection, testing and the disposition of components for manufacturing. Typically, a manufacturing site has endotoxin specifications for all raw materials used in the manufacturing process. Incoming goods are either tested to ensure they conform to these specifications or vendor data found on the Certificate of Analysis/Conformity is used to verify the components pass the site specification. Site procedures can be established such that only components which pass a lower internal endotoxin specification are used in the manufacturing of any final drug product requiring low endotoxin. As the actual endotoxin results during the testing of incoming goods are often much lower than the component specifications, the establishment of site controls for the disposition of only those lots of components which pass a tight specification is usually easily manageable as most incoming goods lots will pass these specifications.
Establishing additional control points at the vendor is another potential way to control the overall endotoxin risk. This is particularly true for process components such as tubing and disposable mixing bags as well as stoppers used in the primary container closure. It is critical to understand how endotoxin is tested at the vendor and whether there are any endotoxin reduction steps, such as additional washing or gamma irradiation, during the manufacturing of these components. Furthermore, the validation status of these endotoxin reduction steps should be assessed as these steps often result in a decrease of endotoxin of at least three orders of magnitude. This results in components that routinely have endotoxin levels significantly below their specification. By sourcing process materials with validated endotoxin reduction steps during their manufacturing, the overall risk of endotoxin to the final drug product can be further mitigated.
Additional in-process endotoxin controls can be implemented into the manufacturing process to further control endotoxin reduction. For example, if the internal endotoxin specification for the WFI line to be released by the manufacturing site is at a value that results in WFI being a high or medium risk component, additional endotoxin testing can be added as part of the manufacturing process. Prior to the initiation of the process, a WFI sample can be pulled and tested for endotoxin. The manufacturing process can continue but the commitment of the API can be held until an endotoxin value for the WFI is obtained that passes an in-process specification significantly below that of the site endotoxin specification, typically 15-20% of the total allowable endotoxin. By doing this, the risk of endotoxin from the WFI can be reduced to a low risk component. Recent advances in rapid endotoxin tests make the establishment of in-process controls for endotoxin easier to establish as the manufacturing process is not held for significant time while testing occurs. Similar in-process controls can be established at other points in the manufacturing process, such as during buffer preparation and drug product compounding, if deemed appropriate.
Finally, the historical results from endotoxin testing of the components can be used to in a risk-based manner to determine if additional controls are required. Many components, such as excipients and primary container components, likely have a significant history of testing results at a manufacturing site. Vendors are also likely to have a similarly significant testing history for the components that they are manufacturing. If an extensive history exists, the worst case historical result can be used in the stack tolerance analysis instead of the theoretical maximum amount calculated from the component’s specification. The historical results are often significantly below the endotoxin specification and can result in a substantial reduction in endotoxin risk from that component.
In cases where a significant amount of historical data is not available, other controls may be necessary. For instance, prior to commercialization, a limited number of API batches may have been produced. Thus, while the endotoxin results for these API batches may be significantly below the specification for the API, enough data may not be available to deem that that overall risk from the API can be reduced. Further mitigation could be the establishment of an internal action limit on endotoxin for the API where if a future batch exceeds the action limit, the impact on the final drug product is considered before releasing the material. Alternatively, some quality systems may flag future API batches that have higher endotoxin levels than those previously produced as “out of trend” and would initiate an assessment as to whether the API was fit to use which would include the impact to drug product manufacturing.
Once pertinent controls have been implemented, the stack tolerance analysis can be performed again using new values for maximum endotoxin risk for each component. If the composite value from all components is now under the final drug product specification, control of endotoxin during the manufacturing process can be established. If the final value is still higher than the specification, further mitigations should be considered.
Conclusion
When developing a drug product with tight endotoxin specifications, control beyond those normally used during the manufacturing process may need to be implemented. In order to establish these controls, it is critical to consider the end-to-end manufacturing process in addition to the risk posed by each individual manufacturing step and component. When considering only individual steps and components, most of these will be classified as moderate or low risk and may be deemed as having acceptable risk. However, by collectively considering all these steps and components in an end-to-end assessment, the composite risk posed by the manufacturing process may exceed the endotoxin allowable per the specification. As such, the end-to-end assessment is critical to identifying the appropriate control points, introducing additional mitigation actions and developing an overall control strategy that results in a manufacturing process that reliably delivers a drug product that will meet specifications.
References
1. United States Pharmacopeial <85>. “Bacterial Endotoxins Test” (2011)
2. United States Pharmacopeial <1211>. “Sterilization and Sterility Assurance of Compendial Articles” (2011)
3. European Pharmocopeia 9.1 Section 5.1.1 “Methods of Preparation of Sterile Products” (2017)
4. Hecker, W., Witthauer, D. and Staerk, A. “Validation of Dry Heat Inactivation of Bacterial Endotoxins”. PDA Journal of Pharmaceutical Science and Technology. Vol 48 (4) 197-204 (1994).
Author Biographies
Dr. Robert Simler is an Associate Director in the Engineering and Technology department at Biogen (Cambridge, MA). He obtained his Ph.D. from the Pennsylvania State University in Chemistry, specializing in protein folding thermodynamics and kinetics. He has 13 years of experience in drug product formulation, process development and biophysics. He has participated in the development of antibodies, glycoproteins, nucleic acids and fusion proteins. He has been at Biogen since 2013 where he leads the antisense oligonucleotide drug product formulation and process development group.
Dr. Brian Thome is a Senior Engineer III in the Parenteral Manufacturing Sciences department at Biogen (Zug, Switzerland). He obtained his Ph.D. in Chemical Engineering from Washington State University specializing in chemical separations and bioprocessing. He has 10 years of experience in drug product technology transfer, fill-finish process development and formulation science and has worked on antibodies, nucleic acids, fusion proteins and blood products. He has been at Biogen since 2010 in a variety of drug product technical roles and now leads the parenteral product operations support team within manufacturing sciences.
Subscribe to our FREE newsletter and WEBINAR UPDATES
We will not sell or give your information to a third party. See our Privacy Policy















